



Entropía
De Laplace
Contenido |
1 Introducción
2 Desigualdad de Clausius
La desigualdad de Clausius establece que, para todo proceso cíclico
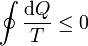
donde cada uno de los símbolos posee la siguiente interpretación
-
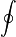
- La integral con el circulito se denomina “integral cerrada” y quiere decir que la suma se efectúa sobre una curva que se cierra sobre sí misma
- dQ
- representa la cantidad de calor diferencial que entra en el sistema desde un foco situado a la temperatura T. A lo largo de un ciclo habrá ocasiones en que su valor sea positivo y veces en que será negativo, según el sistema absorba o ceda calor.
- T
- es la temperatura del foco que cede el calor. No es la temperatura del sistema. Es más, para empezar la temperatura del sistema probablemente ni estará definida. En algunos puntos tendrá un valor y en otros será distinto. En el caso de que sí tenga un valor definido, T', este valor será menor que el exterior cuando el calor entra (ya que si no, no entraría), y será mayor que el exterior cuando el calor sale. Solo en un proceso reversible T' se diferenciará una cantidad infinitesimal de T (ya que si no, no sería reversible).
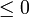
- La desigualdad de Clausius no nos dice cuanto vale la integral, en general. Solo nos informa de sus signo. Pero al hacerlo nos proporciona un criterio para clasificar los posibles procesos:
- Si la integral es negativa: el proceso es irreversible.
- Si la integral es nula: el proceso es reversible.
- Si la integral es positiva: el proceso es imposible.
3 Definición de entropía
3.1 Variación de entropía
Para un ciclo reversible, la desigualdad de Clausius se transforma en una igualdad.
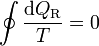 (ciclo reversible)
(ciclo reversible)Consideremos ahora un ciclo reversible que pasa por dos estados A y B. Podemos suponer este ciclo como formado por dos caminos C y C' que van de A a B, siendo uno de ellos (C') recorrido de B a A. Para este ciclo la igualdad se convierte en
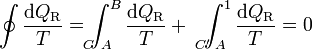
y, despejando

Ahora bien, por ser los caminos reversibles, la integral de B a A por C' es igual a la integral de A a B por el mismo camino C', cambiada de signo. Físicamente, esto quiere decir que si vamos de B a A por C' y en un cierto paso entra en el sistema una cantidad de calor dQR, si recorremos el camino en sentido contrario, cuando lleguemos al mismo sitio la misma cantidad de calor saldrá del sistema (es decir, que en el camino inverso entra − dQR). Por tanto
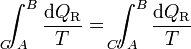
Puesto que este resultado puede extenderse a cualquier otro camino reversible que conecte A con B, concluimos que el valor de la integral es independiente del camino y por tanto solo depende de los estados inicial y final. Por ello, su valor es igual a la diferencia de una cierta función de estado que denominamos entropía
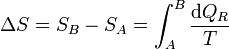
o, en forma diferencial
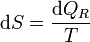
Dicho con palabras: el incremento diferencial de entropía entre dos estados vecinos es igual la cantidad de calor que entra reversiblemente entre dichos estados dividida por la temperatura a la que se intercambia el calor. Puesto que estamos hablando de procesos reversibles no es necesario distinguir entre la temperatura del ambiente T y la del sistema T', ya que en un proceso reversible ambas deben diferenciarse como mucho en una cantidad infinitesimal.
La entropía cumple que:
- Sus unidades en el sistema internacional son el J/K.
- Es una propiedad extensiva, proporcional a la cantidad de masa. Puede definirse la entropía específica, s por unidad de masa, por mol o por unidad de volumen. En cualquiera de estos casos, s es una propiedad intensiva.
3.2 Entropía absoluta
La definición anterior sólo nos da la diferencia de entropía entre dos estados, no su valor absoluto. Es más, hemos afirmado, sin probarlo, que el resultado de la integral es una diferencia de los valores de la entropía en B y en A. ¿Por qué la diferencia y no, por ejemplo, el cociente f(B) / f(A)?
Podemos definir la entropía respecto a un cierto estado de referencia, O, como la integral
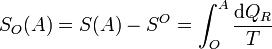
Este estado O, por definición, tendrá entropía nula respecto a sí mismo (SO(O) = 0). Puesto que O es un estado fijo, esta integral solo depende de A.
Consideremos ahora la integral entre A y B
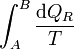
Puesto que podemos elegir el camino que queramos para ir de A a B, tomamos uno que pase por O. Entonces
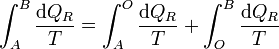
y puesto que los caminos son reversibles
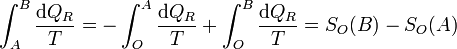
Luego efectivamente sí resulta la diferencia entre la función evaluada en B y la misma función evaluada en A.
Realmente esta definición lo que nos da es de nuevo una diferencia, pero respecto a un estado estándar, que puede ser establecido por convenio. La IUPAC recomienda definir el estado estándar como el correspondiente a una presión de 100 kPa (0.9869 atm) y 0°C (aunque la mayoría de las tablas dan la entropía del estado estandar a 25°C).
El cambio de estado de referencia lo que añade es una constante a la entropía. Por ello, si lo que queremos calcular es la variación de entropía entre dos estados A y B, nos es indiferente el estado de referencia que tomemos, nos basta con hacer la integral directamente por un camino reversible que vaya de A a B.
Cuando se formula el Tercer Principio de la termodinámica, que establece que la entropía de un cristal perfecto, a 0 K es nula, se da un verdadero valor absoluto a la entropía, pues fija un estado de referencia universal.
4 Principio del aumento de entropía
4.1 Ciclo parcialmente irreversible
Supongamos ahora un ciclo irreversible formado por un camino irreversible que lleva del estado A al B y vuelve por un camino reversible. En este caso tenemos la desigualdad
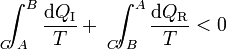
Cambiando uno de los términos de miembro
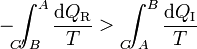
A la integral sobre el camino reversible podemos invertirla y cambiarle el signo (cosa que no podemos con el irreversible, precisamente por ser irreversible). Por tanto la desigualdad anterior equivale a decir que la integral por el camino reversible es mayor que por el irreversible
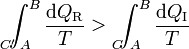
Pero la integral por el camino reversible es justamente la diferencia entre la entropía inicial y la final. Por tanto
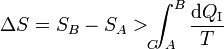
Combinando este resultado con el del caso reversible obtenemos la relación general
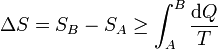
La variación de la entropía es siempre mayor o igual que la integral del calor que entra en el sistema dividido por la temperatura a la que entra. La igualdad se dará en un proceso reversible y la desigualdad en uno irreversible.
4.2 Principio del aumento de entropía
En el caso particular de un sistema aislado, ningún calor entra o sale del sistema, dQ = 0 y por tanto
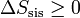 (sistema aislado)
(sistema aislado)esto es, para un sistema aislado la entropía es una función creciente en todo proceso real. Sólo si el proceso es reversible la entropía permanece constante.
Si consideramos el “universo” como el conjunto del sistema más la parte del ambiente con la que intercambia calor o trabajo, podemos tratar al universo como un sistema aislado, con lo que obtenemos el principio del aumento de entropía:
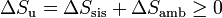
esto es, en todo proceso real la entropía aumenta (si el proceso es irreversible) o permanece estacionaria (si es reversible), pero nunca disminuye. El criterio general es

Obsérvese que en un proceso real es posible que la entropía del sistema disminuya (es lo que ocurre normalmente si se enfría), pero esa disminución debe ser compensada por un aumento en el ambiente, que supere con creces dicha disminución.
5 Producción de entropía
La desigualdad de Clausius en términos de la entropía
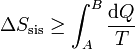
puede convertirse en una igualdad introduciendo un nuevo término
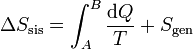
donde Sgen es la producción de entropía. Representa la cantidad de entropía producida en el sistema como consecuencia de las irreversibilidades internas. La desigualdad de Clausius establece, por tanto, el criterio

Escrita la desigualdad de esta forma podemos entender la variación de entropía como suma de dos términos: lo que entra por las paredes debido al calor intercambiado con el ambiente más lo que se produce en el propio sistema.
Otra forma de interpretarlo es escribiendo la igualdad anterior como
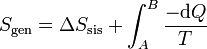
Como se ve más abajo, la variación de entropía de un foco es Q / T, siendo Q el calor que entra en el foco. En la integral anterior − dQ es la cantidad de calor que sale del sistema y por tanto entra en el foco a temperatura T. Al sumar para todos los focos estamos calculando la variación total de entropía del ambiente, Nos queda entonces
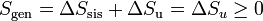
con lo que la entropía generada en el sistema y la variación de entropía del universo son cantidades equivalentes.
6 Variación de entropía en una máquina térmica
6.1 Variación de entropía de un foco térmico
En el caso particular de un foco térmico su capacidad calorífica es tan grande que la entrada o salida en él no produce un aunmento o disminución de su temperatura. Esto quiere decir que el intercambio de calor con un foco es siempre un proceso reversible (desde el punto de vista del foco), por lo que la variación de entropía del foco es simplemente
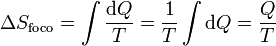
Aquí Q es el calor que entra en el foco (que normalmente saldrá del sistema).
6.2 Variación en un ciclo de la máquina
Cuando tenemos una máquina térmica que intercambia calor con dos focos a temperatura Tc y Tf, la variación de entropía del universo será la suma de la de la máquina más la del ambiente.
6.2.1 Del sistema
La máquina realiza un proceso cíclico. Puesto que la entropía es una función de estado su valor al final del ciclo será el mismo que al principio. Por tanto
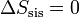 (proceso cíclico)
(proceso cíclico)6.2.2 Del ambiente
La máquina intercambia calor con dos focos térmicos. Del foco a Tc sale una cantidad de calor | Qc | . En el foco a Tf entra un calor | Qf | . Por tanto la variación de entropía del ambiente es
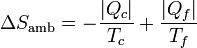
6.2.3 Del universo
Puesto que la entropía del sistema no cambia en un ciclo, la variación de entropía del universo coincide con la del ambiente
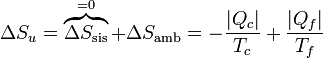
Para que esta máquina pueda operar, esta variación de entropía debe ser positiva. La reducción de entropía del primer foco debe ser compensada con creces por el aumento en el otro. Este criterio limita la eficiencia de máquinas térmicas y refrigeradores.
7 Entropía y trabajo perdido
7.1 Caso de una máquina térmica

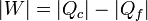
Por lo lado, la producción de entropía, según acabamos de ver es
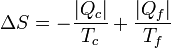
De aquí podemos despejar el calor entregado al foco frío
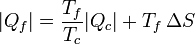
Esta ecuación nos dice que
- Existe siempre un calor entregado al foco frío, esto es, se verifica el enunciado de Kelvin-Planck.
- Para una cantidad fija que se toma del foco caliente, si se genera entropía, es decir, si la máquina es irreversible, la cantidad de calor de desecho es mayor que si no se generara. Dicho en otras palabras, se desperdicia más energía, pues una vez que va a parar al foco frío (normalmente el ambiente que nos rodea) ya no es útil para producir trabajo adicional.
Sustituyendo este calor en la expresión del trabajo obtenemos la relación
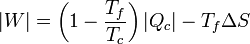
La cantidad entre paréntesis es el rendimiento de una máquina reversible que opere entre las temperaturas Tc y Tc. Por ello, esta ecuación se puede escribir
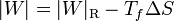
que nos dice que una máquina irreversible produce menos trabajo que una reversible, porque una parte del calor se desperdicia de forma irrecuperable. Este trabajo perdido es proporcional a la entropía creada
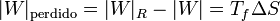
Cuanto más entropía estemos generando, más trabajo potencial se pierde y menos produce la máquina.
En términos del rendimiento, podemos escribir la ecuación anterior como
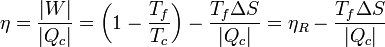
lo que nos expresa el teorema de Carnot: el máximo rendimiento lo obtenemos con una máquina reversible, y a partir de ahí empezamos a perder eficiencia, proporcionalmente a la entropía generada (para una entrada de calor siempre la misma).
También pueden expresarse estos resultados en términos del trabajo que queremos obtener de la máquina
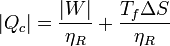
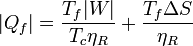
que nos dice que, si queremos obtener un trabajo dado, cuanto más irreversible sea la máquina más calor necesitamos tomar del foco caliente y más calor de desecho arrojamos al foco frío.
7.2 Caso de un refrigerador

Un refrigerador extrae un cierto calor | Qf | a una temperatura Tf, para lo cual necesita realizar una cierta cantidad de trabajo | W | . El refrigerador entrega una cantidad de calor de desecho | Qc | a un foco caliente (normalmente el ambiente) a temperatura Tc. Una bomba de calor es exactamente lo mismo, salvo que el ambiente es el foco frío y lo que interesa es el calor que entrega al foco caliente. El Primer Pincipio de la termodinámica nos dice que
Por lo lado, la producción de entropía corresponde a que extraemos | Qf | del foco frío y entregamos | Qc | al caliente:
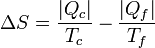
Despejando el calor entregado al foco caliente
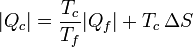
Esta ecuación nos dice que para una cantidad fija que se extrae del foco frío,la generación de entropía por el refrigerador incrementa la cantidad de calor de desecho. Dicho en otras palabras, se desperdicia más energía, pues una vez que va a parar al ambiente que nos rodea) ya no es útil para producir trabajo adicional.
Sustituyendo en la expresión del trabajo
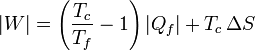
La cantidad entre paréntesis es la inversa del coeficiente de desempeño de un refrigerador reversible que opere entre las temperaturas Tc y Tc. Por ello, esta ecuación se puede escribir
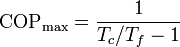
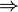
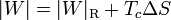
que nos dice que un refrigerador irreversible requiere más trabajo para extraer la misma cantidad de calor y que ese trabajo extra se desperdicia como calor de desecho. El trabajo extra es proporcional a la entropía creada
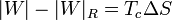
En términos del coeficiente de desempeño, podemos escribir la ecuación anterior como
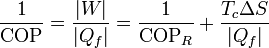
lo que nos expresa el teorema de Carnot para refrigeradores: el máximo coeficiente de desempeño lo alcanzamos con un refrigerador reversible, y a partir de ahí empezamos a perder eficiencia, más cuanto más entropía se produzca (para un calor extraído constante).
También pueden expresarse estos resultados en función del trabajo necesario
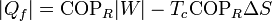
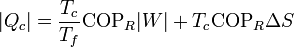
que nos dice que, para un trabajo dado, cuanto más irreversible sea el refrigerador menos calor extraemos del foco caliente y más calor de desecho arrojamos al foco caliente.
8 Ejemplos
8.1 Cambio de fase
Un cambio de fase es un proceso aproximadamente isotermo, port lo que la variación de la entropía de una sustancia cuando experimenta un cambio de fase es simplemente
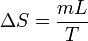
siendo L la entalpía por unidad de masa del cambio de fase (de fusión o de vaporización, por ejemplo, para el agua).
Así, en el paso de hielo sólido a agua a 1 atm se produce un aumento de entropía por unidad de masa (s = S / m):
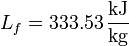
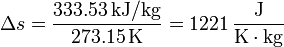
y en el paso de agua a vapor de agua a 1 atm
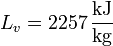
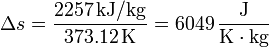
Vemos que es mucho mayor el aumento en la ebullición que en la fusión. Esto está asociado con el gran aumento del desorden al pasar de líquido a gas.
8.2 Calentamiento de una sustancia pura
Cuando se tiene una sustancia pura, como el agua, que experimenta una variación de su temperatura, siendo la presión inicial y la final la misma, la variación de la entropía de la sustancia es, según la definición
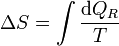
Como proceso reversible podemos imaginar uno en el que la temperatura de la sustancia va variando gradualmente por igual en todos sus puntos, siendo la presión siempre la misma. En este caso
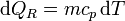
y la variación de entropía es
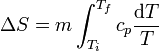
El calor específico es, en general, una función de la temperatura y debe ser tenido en cuenta a la hora de integrar. No obstante, si el rango de variación de la temperatura es pequeño, usualmente se puede aproximar cp por una constante, su valor medio, y obtener la expresión
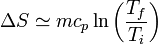
Combinado este resultado con el anterior, podemos trazar la gráfica de la entropía por unidad de masa, como función de la temperatura, para el agua desde el estado de hielo al de vapor:
8.3 Gas ideal
8.4 Mezcla de gases
8.5 Reacciones químicas
Cuando se analiza una reacción química lo que se suele hacer es
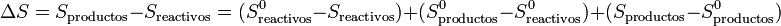
siendo S0 las entropías medidas y tabuladas en el estado estándar (normalmente a 100 kPa y 298 K). La segunda diferencia representa la variación de la entropía en la reacción en el estado estándar
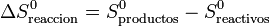
Lo que quiere decir esta descomposición en tres sumas es que, para estudiar una reacción a una cierta presión y temperatura, aprovechamos el que la entropía es una función de estado. En lugar de calcular la diferencia directamente a dicha p y T, lo que hacemos es imaginarnos que llevamos los reactivos, sin reaccionar, al estado estándar. Luego analizamos la reacción en el estado estándar, aprovechando los valores tabulados. Por último llevamos los productos a las condiciones originales.

